AG-0215 Technical Report Series
In-depth scientific analysis of AG-0215's mechanisms, selectivity, and therapeutic potential across multiple indications.
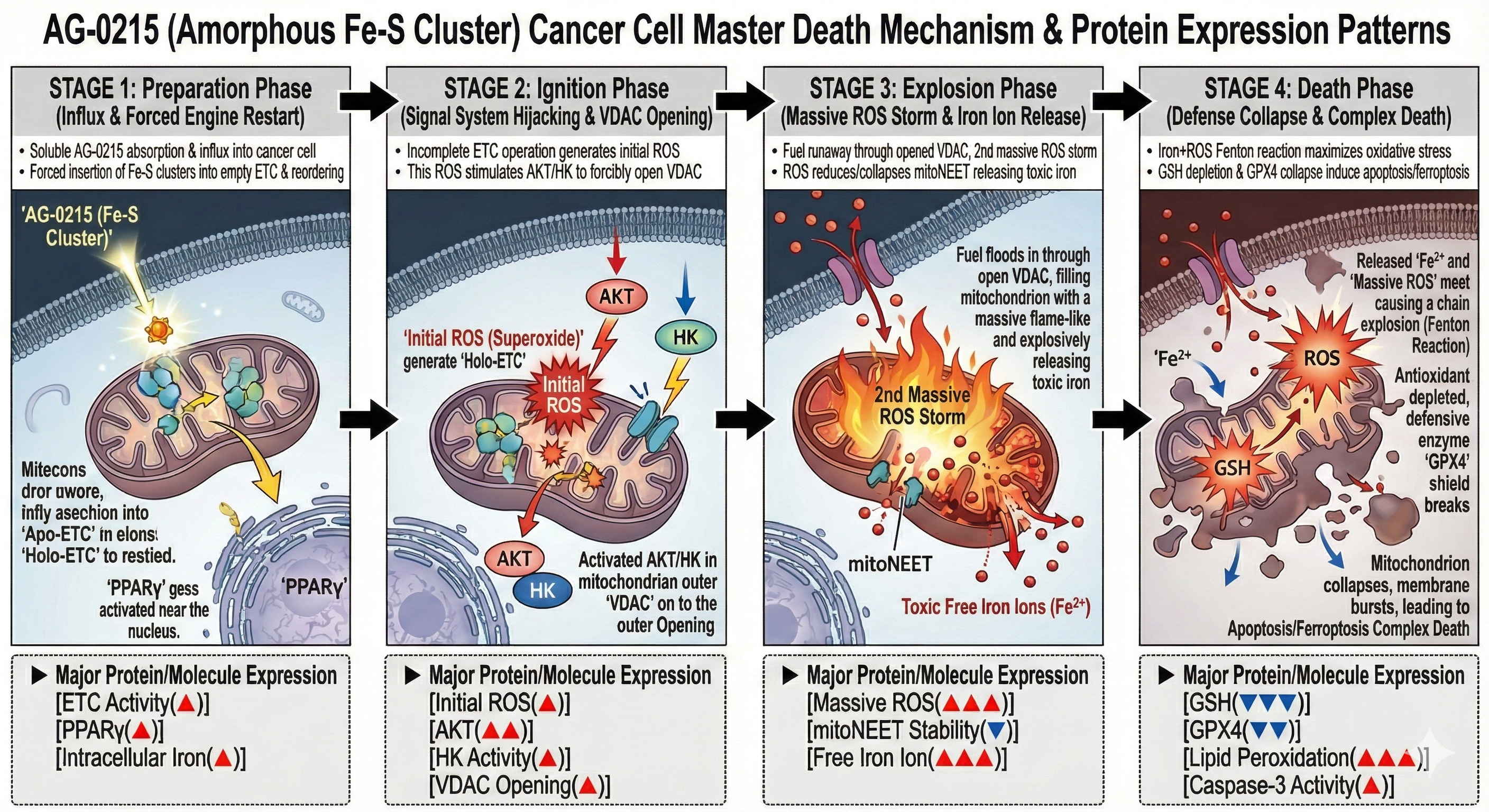
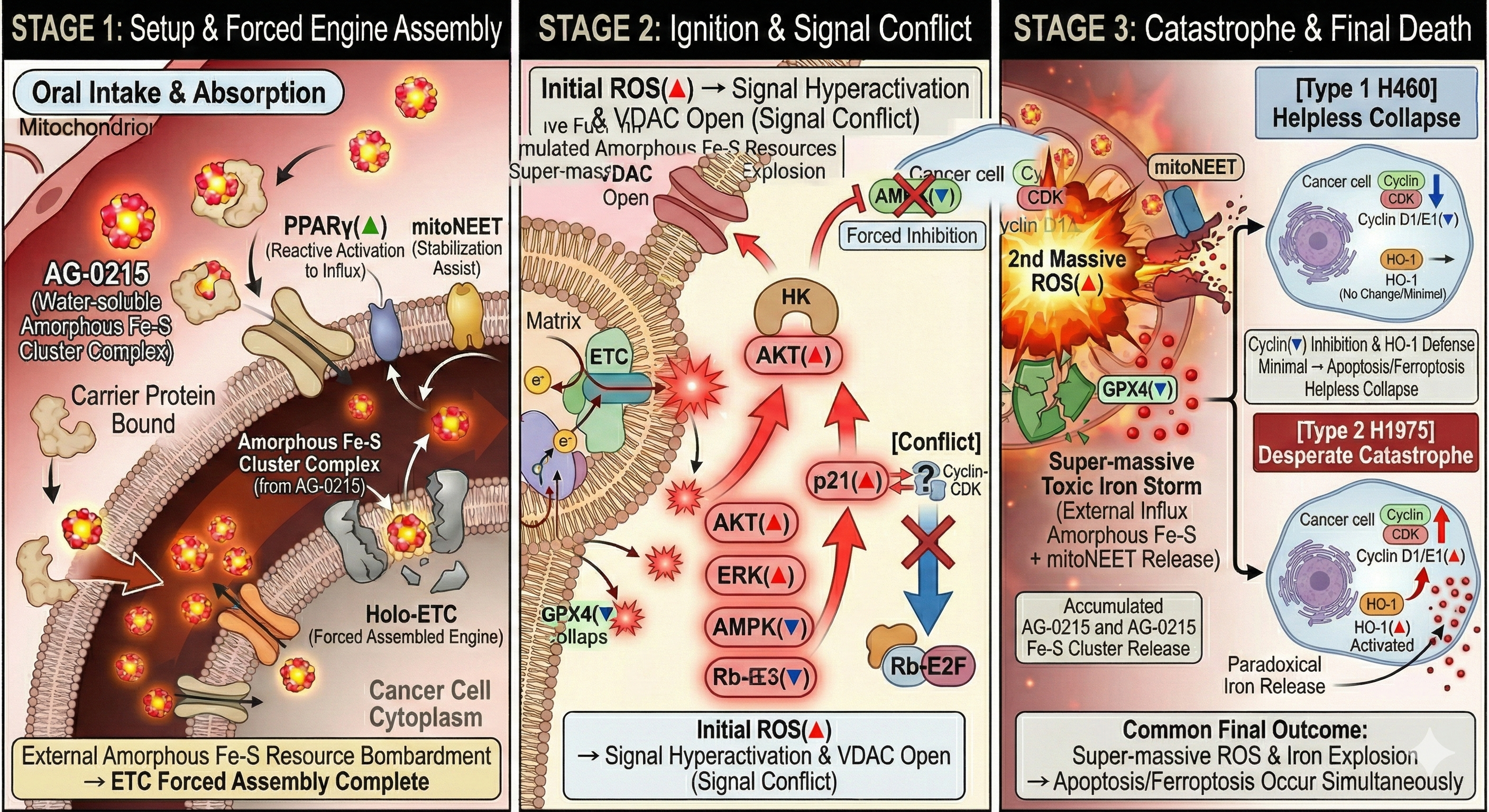
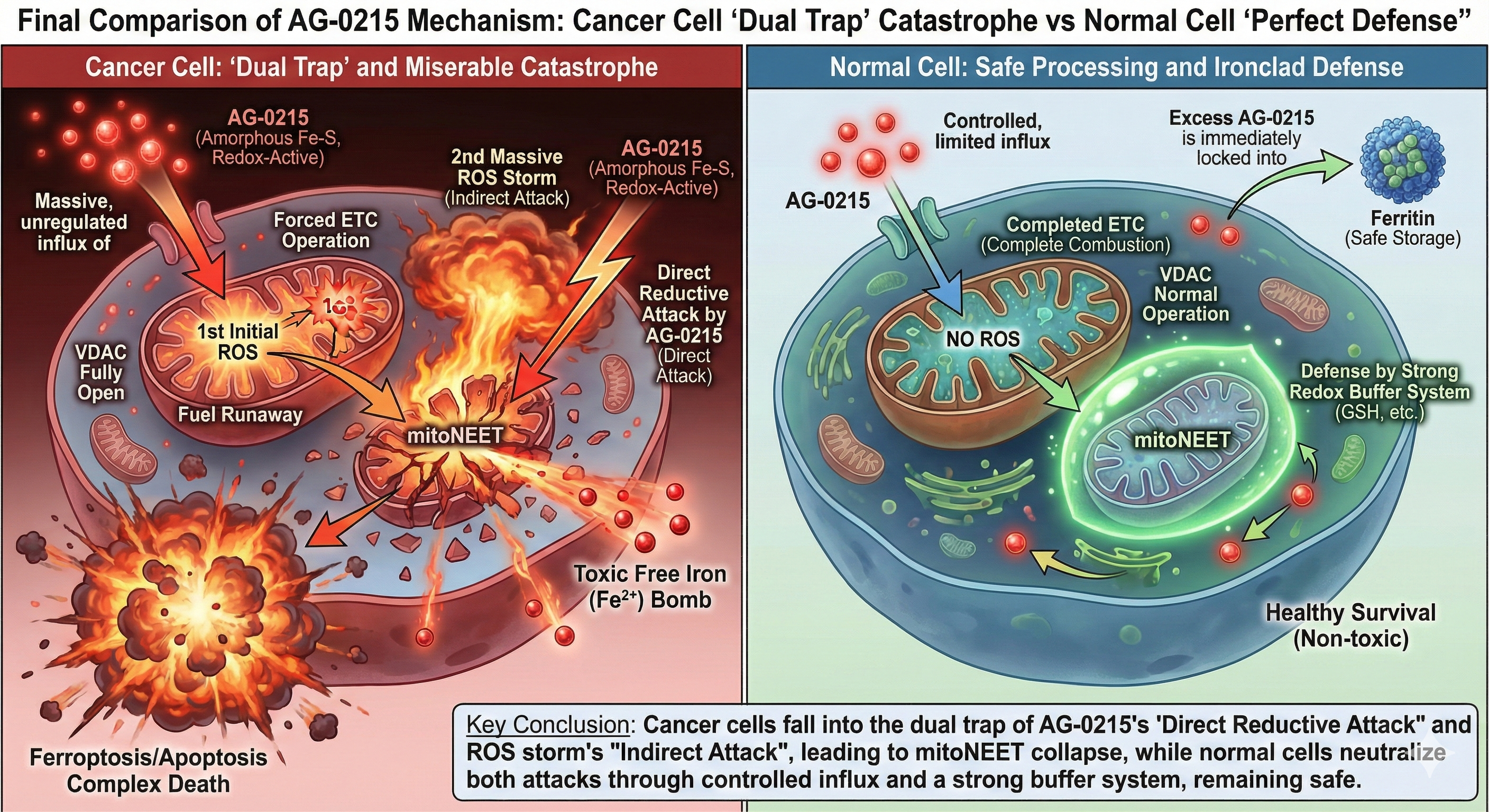
Next-Generation Cancer Cell Death Mechanism (Complex Death)
Analyzes the four-stage mechanism of AG-0215, detailing how the agent exploits mitochondrial vulnerabilities of cancer cells to simultaneously induce apoptosis and ferroptosis via cell-mediated targeted delivery.
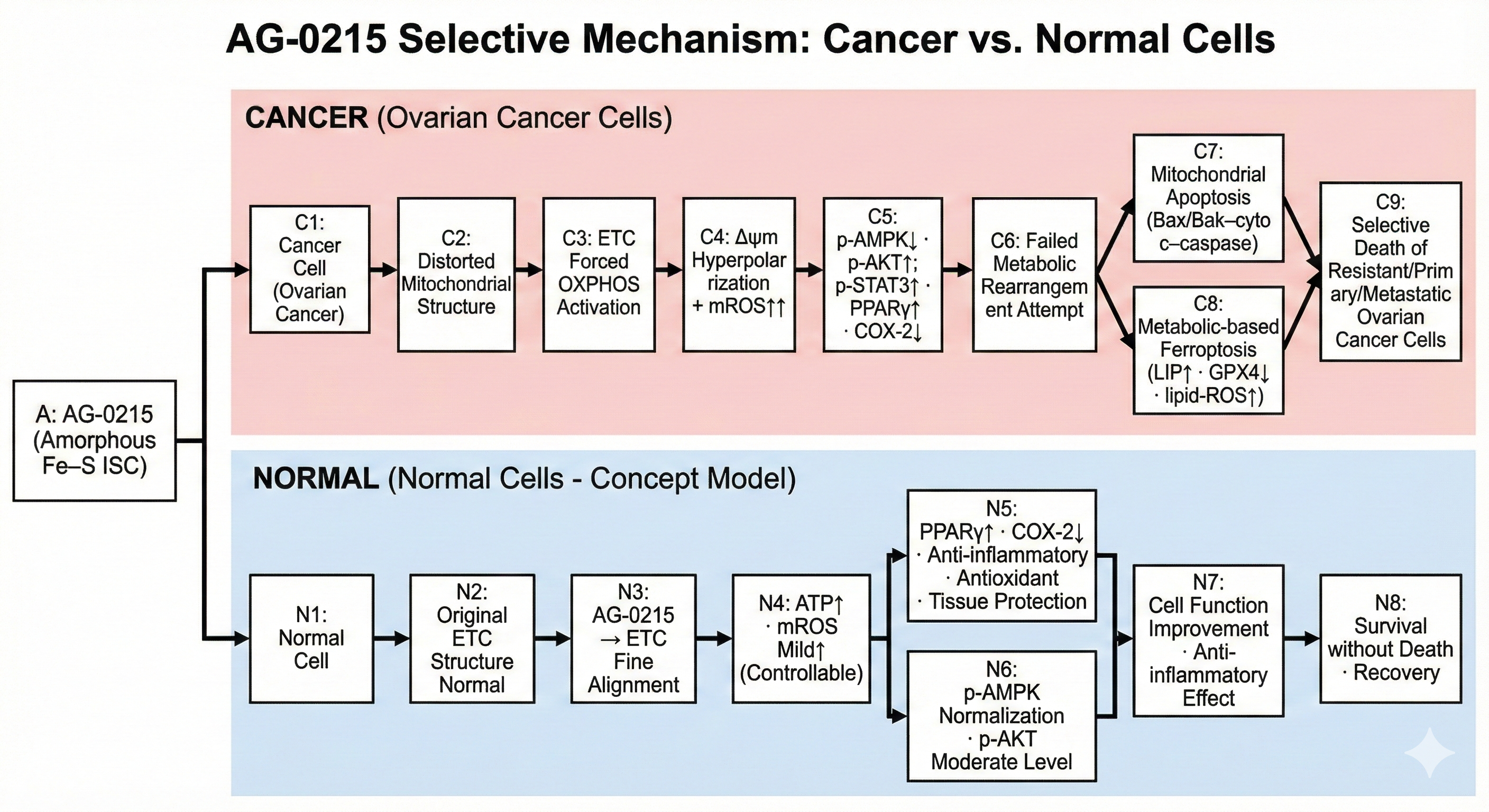
Selective Cancer Cytotoxicity & Normal Cell Bio-Enhancement
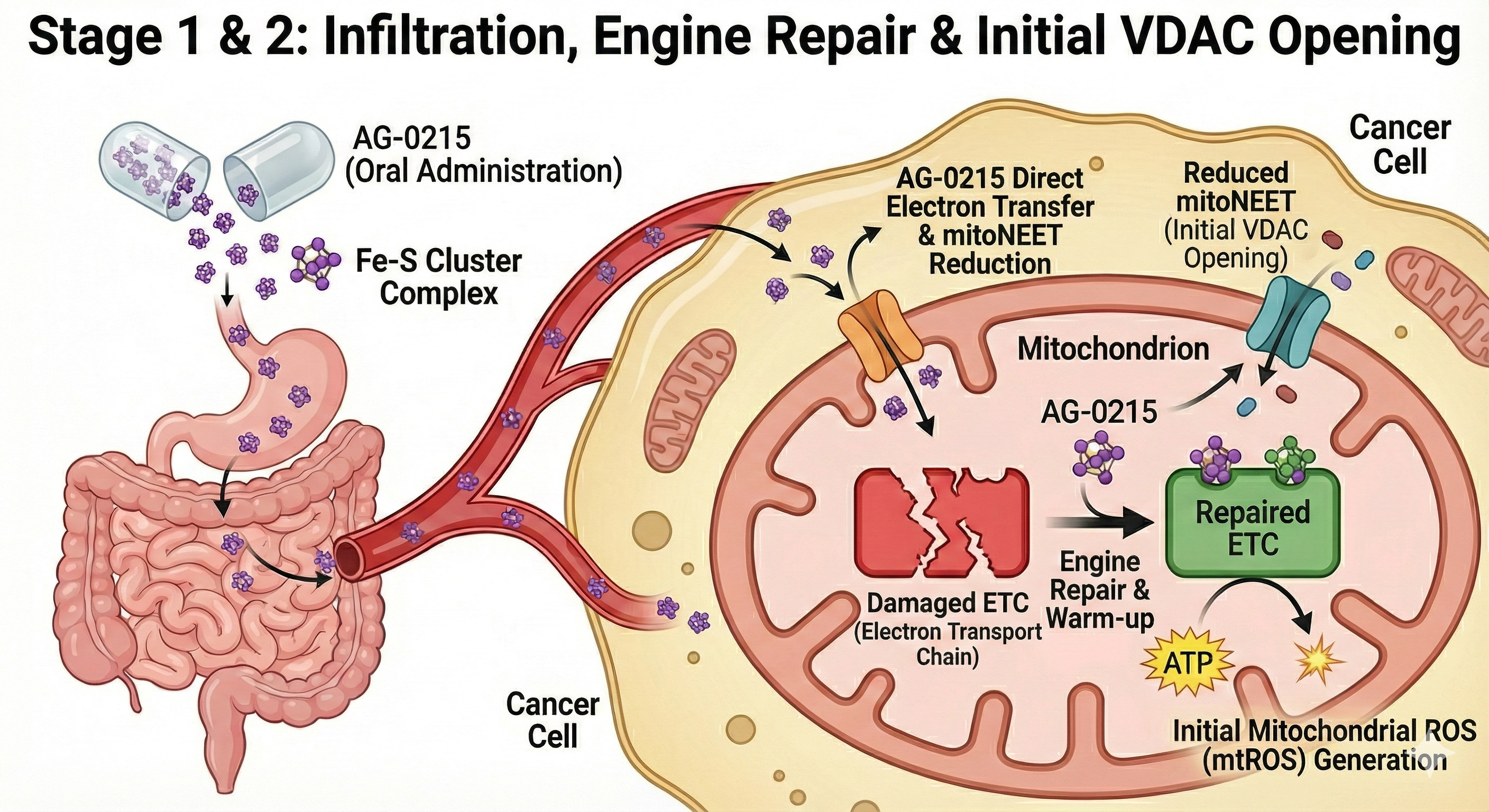
Delineates the bifurcated metabolic pathway of AG-0215 in ovarian cancer. Unlike conventional chemotherapeutics, AG-0215 triggers lethal metabolic collapse in cancer cells while promoting energetic efficiency and anti-inflammatory protection in normal cells.
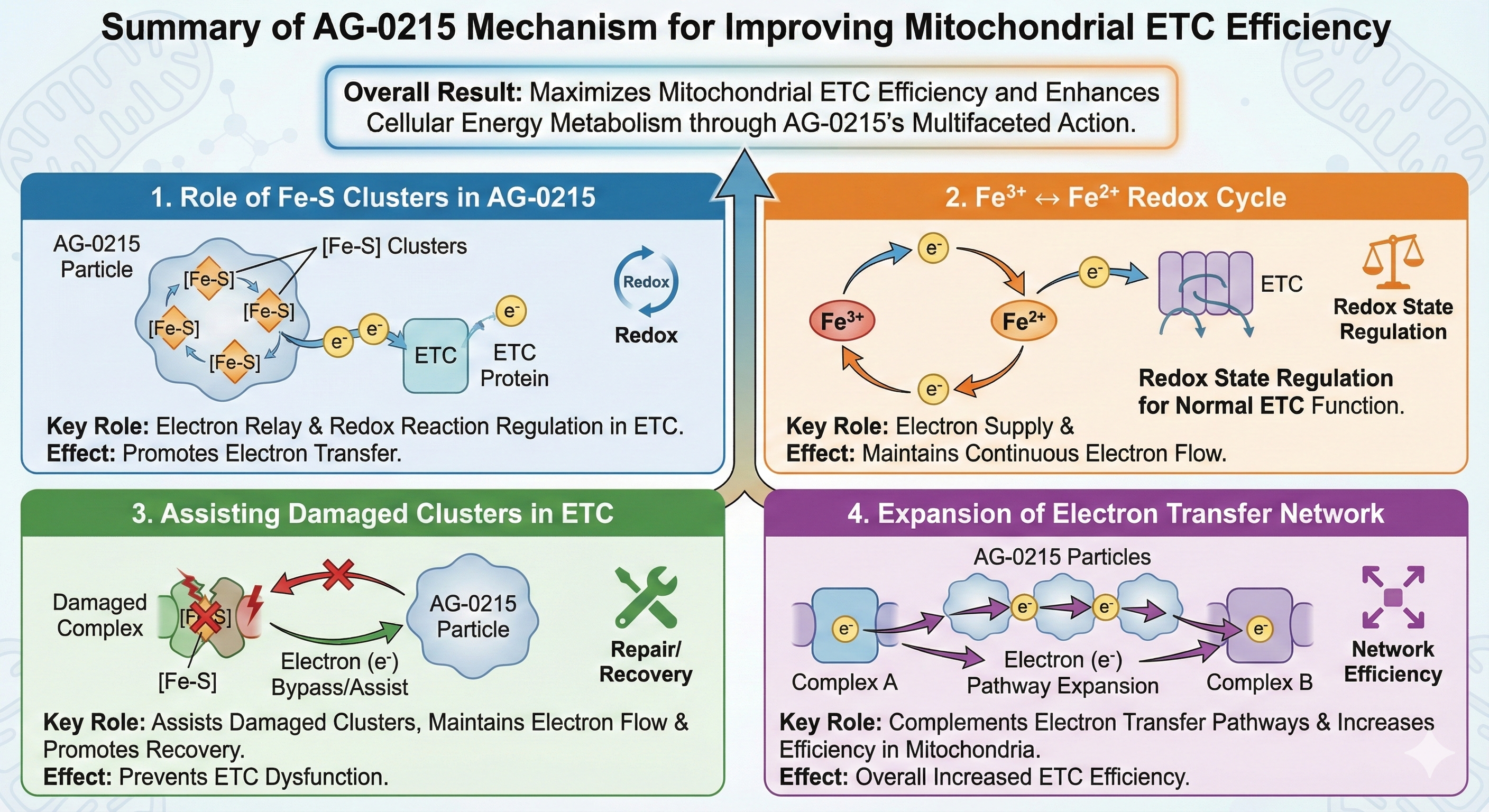
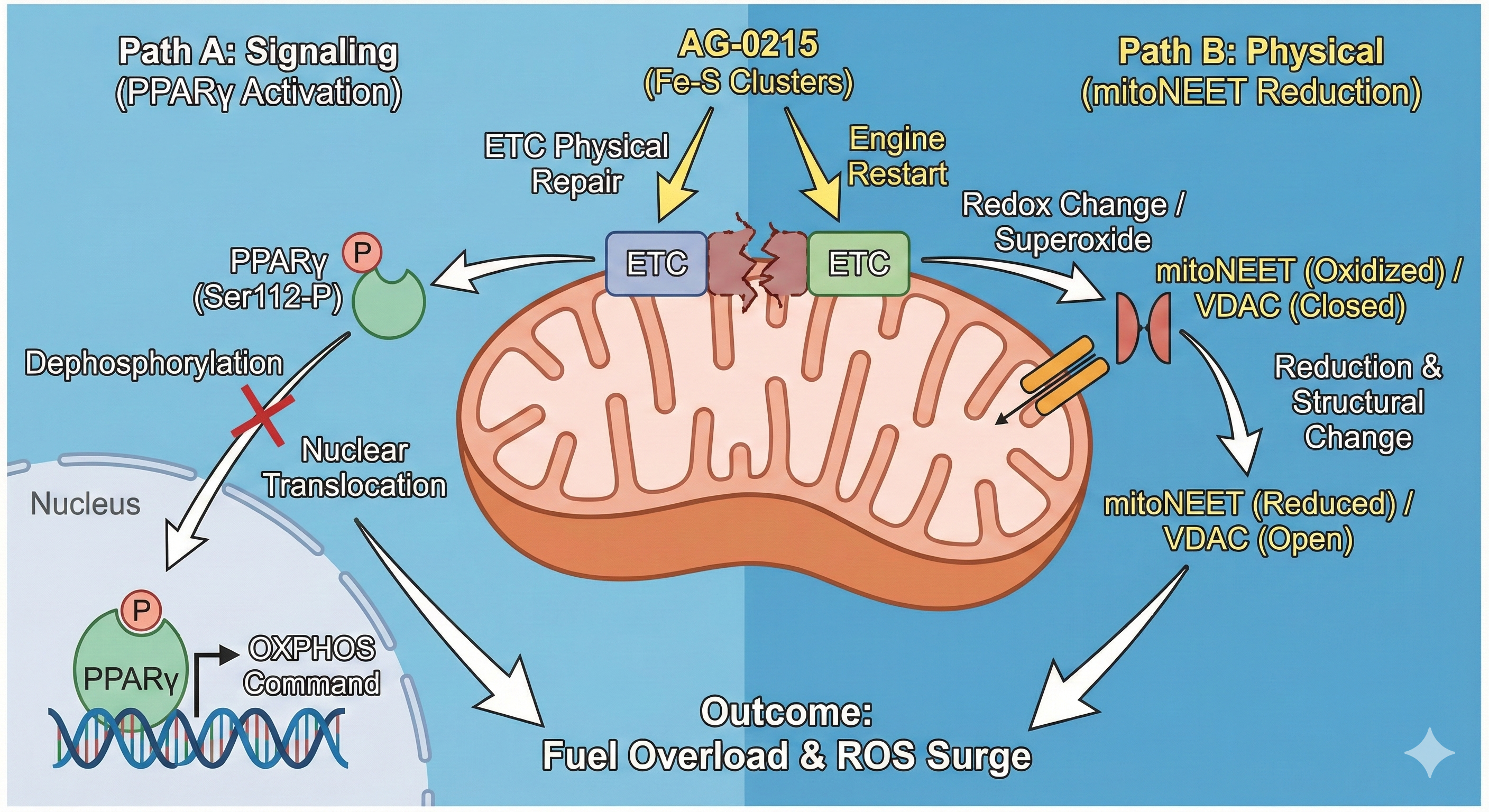
Multifaceted Mechanisms for Maximizing Mitochondrial ETC Efficiency
AG-0215 functions as a bio-catalytic scaffold that facilitates electron flow, mitigates oxidative bottlenecks, and bypasses damaged enzymatic complexes. A paradigm shift from traditional antioxidants that merely scavenge radicals.
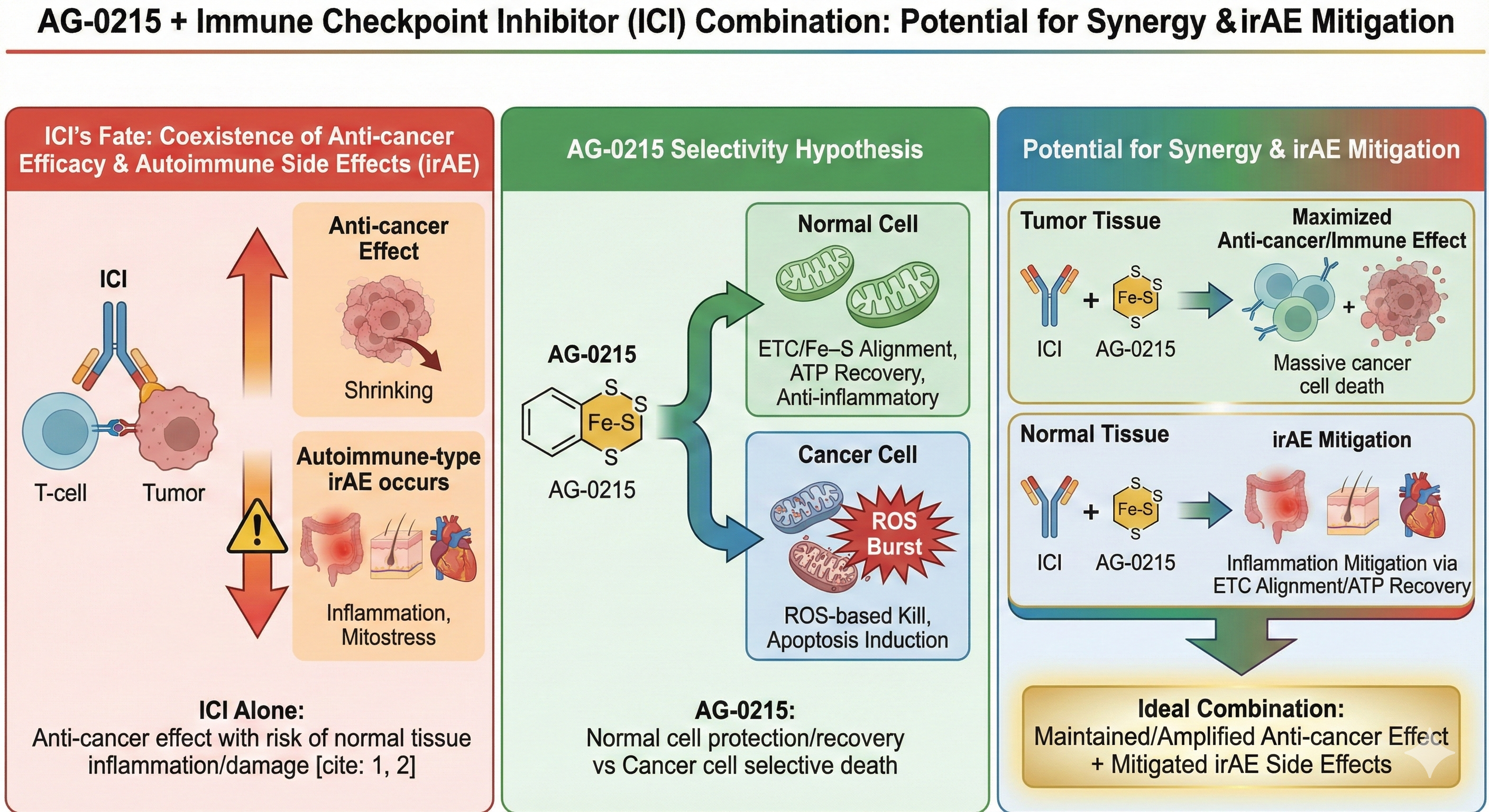
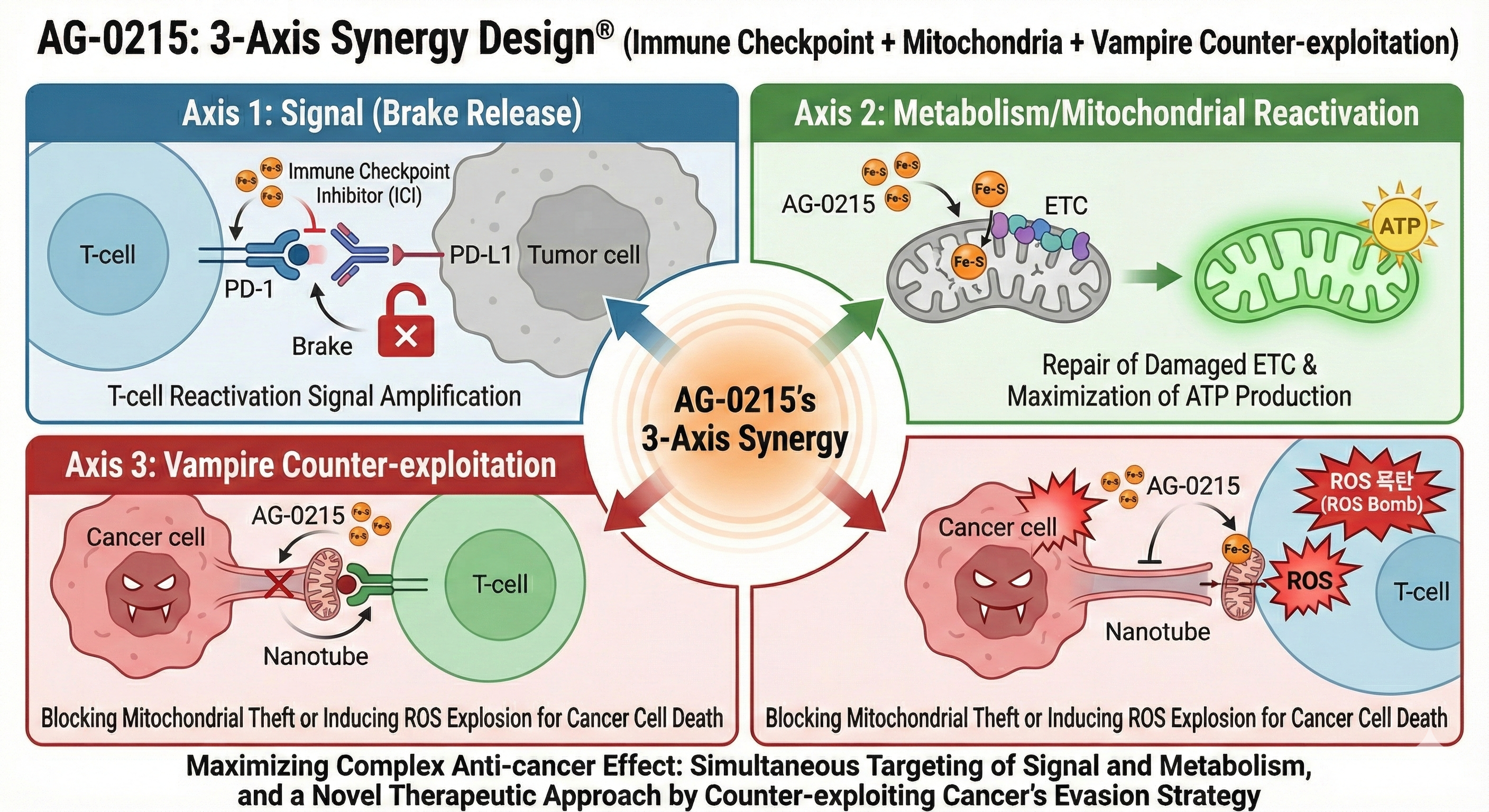
Synergistic Efficacy with Immune Checkpoint Inhibitors (ICI) & irAE Mitigation
Explores AG-0215's potential to selectively amplify anti-cancer efficacy of ICIs while simultaneously mitigating immune-related Adverse Events (irAEs) through mitochondrial metabolic alignment.